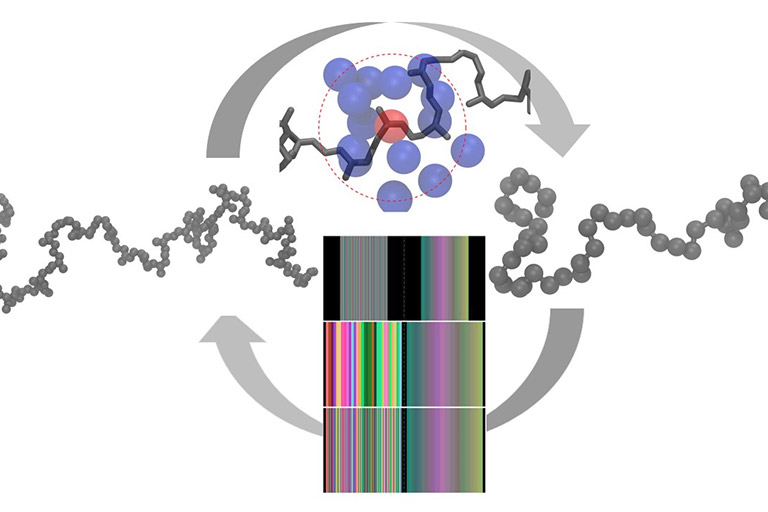
Modeling of commercial-grade polymers in atomistic detail faces challenges due to the requirements of large system sizes and extreme simulation times that are beyond reach of contemporary computational power. Multiscale methods exchange information between coarse and fine representations of molecules to capture material properties over a wide range of spatial and temporal scales. Techniques designed to address this persistent challenge balance among accuracy, efficiency, and general applicability.
Manolis Doxastakis’s group has recently reported on improved methods to design coarse-grained models that systematically match structural parameters of detailed atomistic simulations of polymers. The technique described in the article by Shahidi et al., develops simpler models based on information of the local environment (density) that is passed to an inverse Monte Carlo optimization scheme. This approach holds significant potential to model complex multicomponent systems with simple, efficient and more accurate coarse-grain models.
In a second publication by the group, Li et al. demonstrated that once realistic configurations of long macromolecules have been obtained with the efficient low-resolution model, the missing atomistic information can be introduced with minimal human intervention. Using machine learning algorithms developed originally to enhance image resolution, detailed structures of commercial grade polymers could be proposed closing therefore the loop between atomistic and coarse-grain descriptions.
Read more about the research in The Journal of Chemical Physics Volume 152 Issue 12 and Volume 153 Issue 4.